近日,国科大杭州高等研究院(以下简称“杭高院”)陈洛南研究团队在美国科学院院刊PNAS杂志合作发表题为“Mining Lysine Post-Translational Modification Site by Integrating Protein Language Model Representations with Structural Context”的研究论文。该研究为赖氨酸翻译后修饰位点预测开发了一个整合序列与结构信息的AI框架,可有效发掘PTM位点,促进了PTM相关调控特征的探索,从而为蛋白质调控的进一步研究提供支持。
翻译后修饰(PTMs)是蛋白质功能多样化的关键机制,其中赖氨酸因其高反应活性成为修饰的核心靶点。传统质谱鉴定方法成本高昂且耗时,推动了计算预测方法的发展。当前机器学习模型虽已取得进展,但依赖人工特征工程且缺乏通用性。此外,三维结构信息在PTM位点预测中未能被充分利用。开发集成序列与结构特征的新型预测框架,对于高效发现新型修饰位点并推动实验验证具有重要意义。
研究构建的预测模型创新性地融合了蛋白质语言模型与图神经网络,以同步捕获修饰位点的序列上下文与三维空间结构信息。通过ESM-2提取序列特征,结合基于原子坐标构建的局部接触图进行图卷积处理,最终经多层感知机整合实现高精度预测。
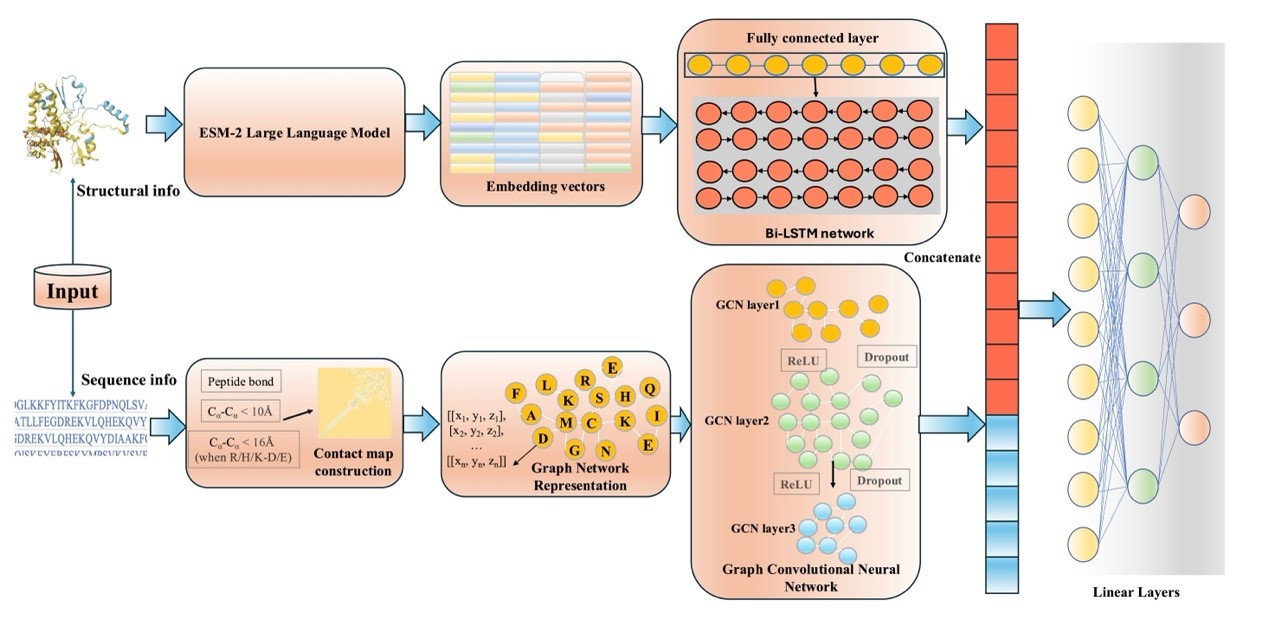
图1 预测模型概览 该模型主要由结构信息处理模块和序列信息处理模块构成。两个模块输出的特征向量经拼接后,通过全连接网络进行降维以生成最终预测结果。具体而言,结构信息处理模块基于氨基酸的原子级三维坐标构建接触图,并利用图神经网络进行处理;序列信息处理模块则通过大语言模型获取表征,再经由线性层和双向长短期记忆网络(Bi-LSTM)进行特征加工。
研究同时采用严格的非冗余评估设置(序列相似度≤40%)与反映真实生物学分布的原始数据集进行双重验证。模型在类别平衡的训练集/验证集上优化,并在高度不平衡的测试集上评估,确保其泛化能力与实用性。结果表明,模型在排除同源偏见和贴近真实场景的条件下均表现出稳健的预测性能。
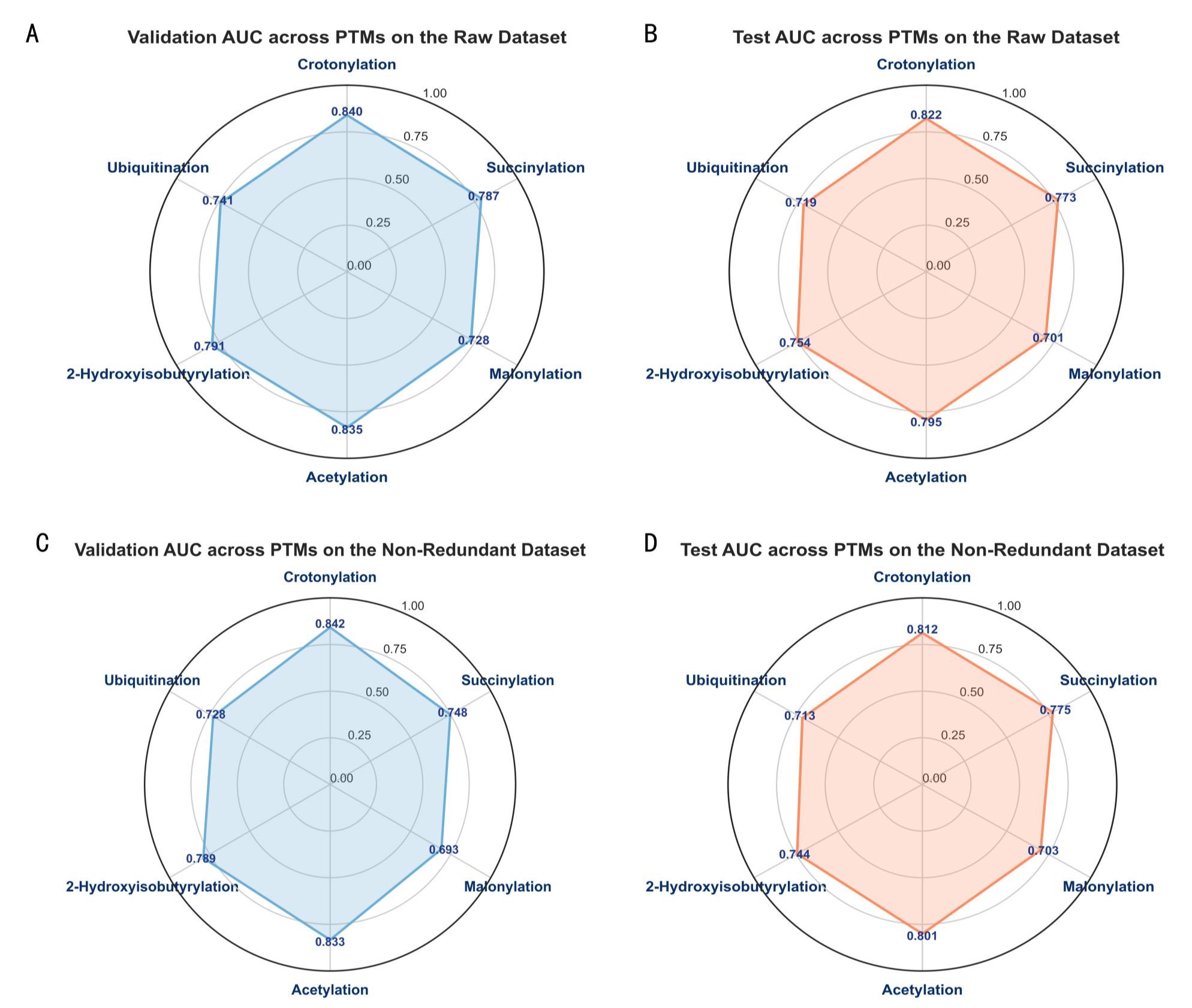
图2 不同数据模式下,模型在六种翻译后修饰类型中的AUC性能表现 A. 原始数据集上各修饰类型的验证集AUC B. 原始数据集上各修饰类型的测试集AUC C. 非冗余数据集上各修饰类型的验证集AUC D. 非冗余数据集上各修饰类型的测试集AUC。
研究在六种代表性翻译后修饰(琥珀酰化、巴豆酰化、丙二酰化、2-羟基异丁酰化、泛素化、乙酰化)的真实生物学分布原始数据上评估了模型的预测能力。采用类别平衡的数据集和严格的蛋白质级划分(训练:测试=7:3),模型经过训练后达到有效拟合。测试集性能显示,模型整体表现稳健,F1分数介于71.7%至80.9%之间,AUC值在78.5%至88.3%范围内。其中,巴豆酰化预测效果最优,F1分数达80.9%,AUC为88.3%;乙酰化与琥珀酰化的F1分数均超过76%,AUC高于83%;而丙二酰化与泛素化的预测性能相对较低但稳定,F1分数约72%。结果重复评估的波动范围较小(±1.0–1.5%),表明模型具有稳定的泛化能力。这些结果验证了框架在多种修饰类型上的有效性与可靠性。
研究将训练好的模型预测了hCLEC12A蛋白的潜在修饰位点(K174与K181),并构建了5种修饰体系进行分子动力学模拟。结合自由能计算显示,相较于未修饰体系(结合能为-58.18 kcal/mol),单一位点修饰(如K181乙酰化)及双修饰均显著削弱了hCLEC12A与抗体50C1的结合(如K174巴豆酰化与K181乙酰化双修饰体系的结合能降至-40.60 kcal/mol)。能量分解分析表明,修饰导致界面关键残基(如K181、R201、R204等)的能量贡献由负转正或大幅降低,破坏了原有的氢键、盐桥等相互作用网络。结果表明,关键赖氨酸的翻译后修饰可通过干扰界面残基间的相互作用显著降低蛋白与抗体的结合亲和力。
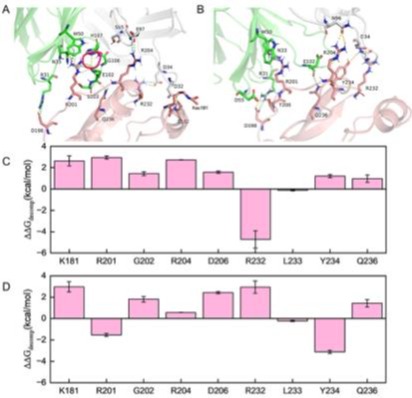
图3 hCLEC12A翻译后修饰对50C1抗体识别的影响。(A) 50C1与hCLEC12A的结合界面示意图:(A) K181乙酰化修饰与(B) K174巴豆酰化与K181乙酰化双修饰情况。黄色虚线、青色虚线与紫色虚线分别代表氢键、盐桥及π-π堆积相互作用。关键残基的单位点结合自由能分解变化:(C) K181乙酰化修饰及(D) K174巴豆酰化与K181乙酰化双修饰体系相较于未修饰纯体系的能量差异(∆∆Gdecomp=∆Gdecomp(修饰体系)-∆∆Gdecomp(纯体系))。数据以均值±标准误表示,数值越小表示该氨基酸对hCLEC12A与50C1结合的贡献越大。
研究提出的融合序列语义与三维结构信息的深度学习通用框架,为精准预测蛋白位点提供了新范式。该框架的模块化设计可扩展至其他残基水平的功能注释任务,推动了计算生物学方法的通用化发展。通过分子动力学模拟验证,框架成功揭示了修饰位点调控蛋白-抗体结合的具体机制,为靶向修饰的疾病干预策略提供了理论依据。
陈洛南研究团队长期围绕生物医学数据挖掘开展系统研究,综合运用深度学习与语言大模型技术,构建面向生物医学的信息自动抽取/预测模型。未来,团队将进一步拓展跨源知识整合与因果关系建模,推动生物医学知识自动化发现与精准预测的发展。
杭高院罗梦奇博士、晨伫科技朱晓红博士为该文章共同第一作者;杭高院/上海交通大学陈洛南博士、南加大/晨伫科技Warshel博士、白晨博士为该文章共同通讯作者。研究工作得到了国家自然科学基金和杭高院的大力支持。
原文链接:www.pnas.org/doi/10.1073/pnas.2529141123
