与大众更熟悉的“吃坏肚子”不同,侵袭性非伤寒沙门菌(invasive non-typhoidal Salmonella, iNTS)一旦突破肠道屏障进入血液,往往引发更加严重的系统性感染,甚至可导致败血症和死亡。世界卫生组织在2025年“大流行病”优先病原体清单中,已将沙门菌列为最重要的五种病原菌之一。因此,iNTS早已不是一个局限于局部胃肠炎的传统感染问题,而是一个日益突出的全球公共卫生挑战。
近年来,杭高院生命学院乐敏课题组持续围绕侵袭性非伤寒沙门菌开展系统研究,从我国流行血清型的识别,到感染致病机制的解析,再到传播链条与跨宿主风险的评估,逐步搭建起对这一类病原体的整体认知框架(Nat Med 2025; J Glob Health 2026; mLife 2024;EMBO Mol Med 2022; Front Microbiol 2021; Microb Genom 2020)。也正是在这些前期工作的基础上,一个更核心更具全球意义的问题浮现出来:作为iNTS中最受关注的高风险分支之一,鼠伤寒沙门菌ST313究竟具有怎样的全球种群结构?它如何跨越大洲传播?又是什么力量在推动其不同谱系此消彼长、不断更替?

为了系统回答上述科学问题,研究整合了迄今规模最大的全球iNTS人源基因组资源,共纳入11,361株人源iNTS基因组以及3,115株ST313基因组,时间跨度覆盖1966年至2023年,为ST313绘制出一张兼具“家谱”与“迁徙图”的高分辨率演化地图。首先ST313究竟主要分布在哪里,又与什么类型的感染最为相关?结果显示,在现有全球样本中,ST313显著集中于非洲,尤其是撒哈拉以南地区;在具有明确来源信息的分离株中,72.7%来自非洲,而在人源样本里,血液分离株高达84%。这意味着,ST313并不是一个普通意义上“容易引起腹泻”的沙门菌分支,而是一个与血流感染高度相关、侵袭性极强的高风险克隆。这样的分布特征,也从流行病学层面再次解释了为什么ST313会长期被视作iNTS研究中的关键对象。
进一步通过全基因组系统发育分析探究其演化变迁,不仅识别出一个高度多重耐药的新亚系L2.4,还定义了一个此前未被充分认识的新谱系L4。尤其是L2.4,携带了包括 blaOXA-1 和 blaCTX-M-15 在内的超广谱β-内酰胺酶相关耐药基因,呈现出明显增强的耐药趋势。换言之,ST313并不是一个相对稳定、已经被认识清楚的旧病原体群体,而是仍在持续分化、持续进化、持续形成新威胁的动态病原群。
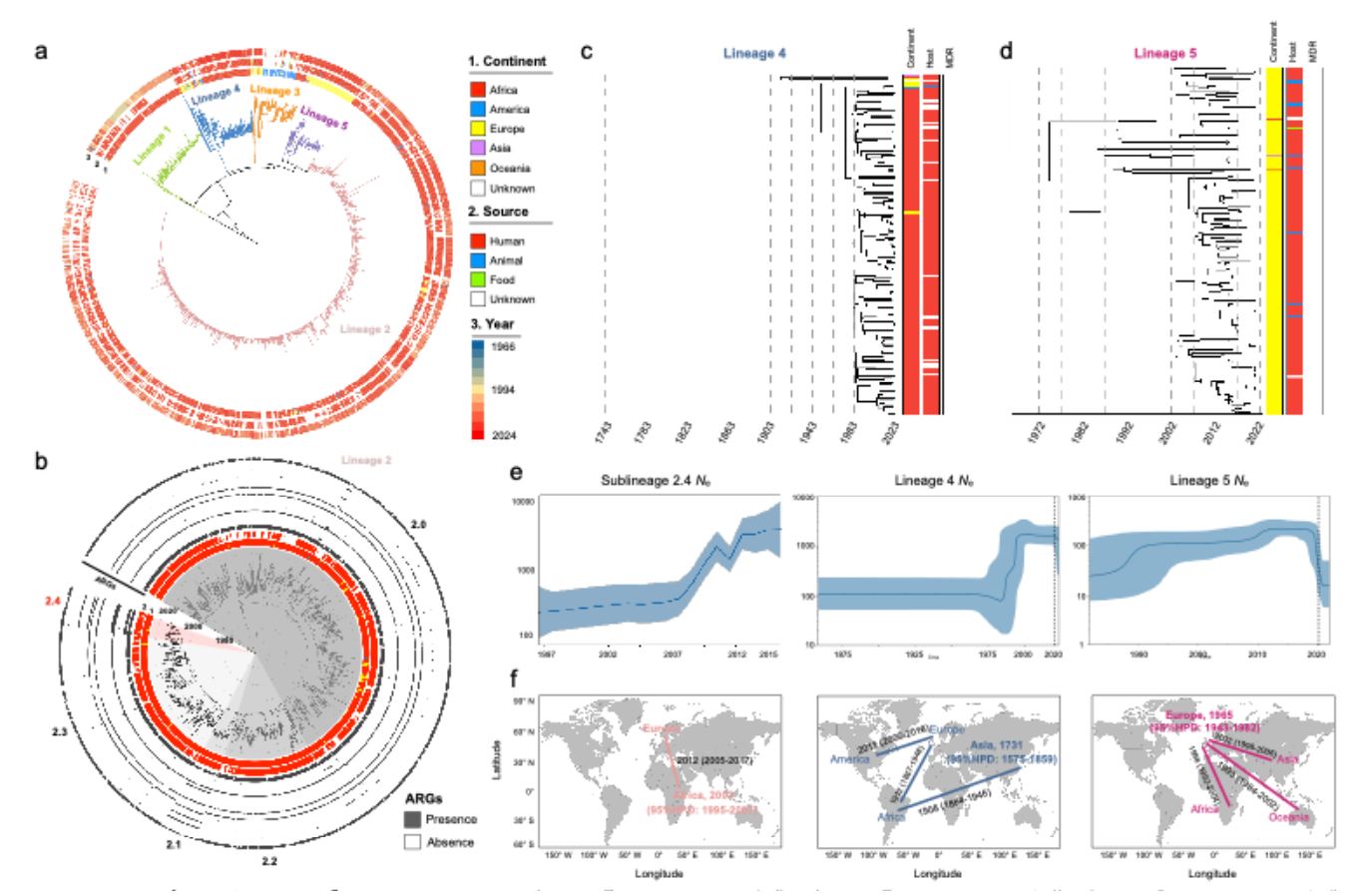
在传播层面,研究结合全基因组系统发育与贝叶斯时空动态分析,重建了ST313主要谱系的传播历史。提示ST313存在清晰的洲际传播事件,而且这种扩散并非孤立发生,而是与现代人口流动和跨区域连接增强相呼应。更重要的是,作者的分析整体支持ST313在人群中的传播链占据重要地位,进一步支撑iNTS菌株以人际传播为主的理论。同时,研究也提示跨宿主传播并未被完全排除,说明这一病原体的传播生态仍然比我们想象得更复杂。
进一步回答了“为什么某些谱系会崛起,而另一些会衰退”这一进化问题。研究显示,推动ST313谱系更替的,并不只是单个耐药基因本身,而是包括质粒、转座子、整合子等在内的可移动遗传元件(mobilome)所驱动的整体耐药基因动态变化。L2.4的崛起与耐药基因的富集密切相关,而L1在非洲的衰退,则与2006年后可移动遗传元件介导的耐药基因携带变化相伴发生。也就是说,ST313不同谱系的命运,在很大程度上是由抗菌药物压力与可移动遗传元件共同塑造的。病原体并不是被动承受外界选择,而是在不断重组、不断试错、不断筛选出更适合当前环境的基因组合。
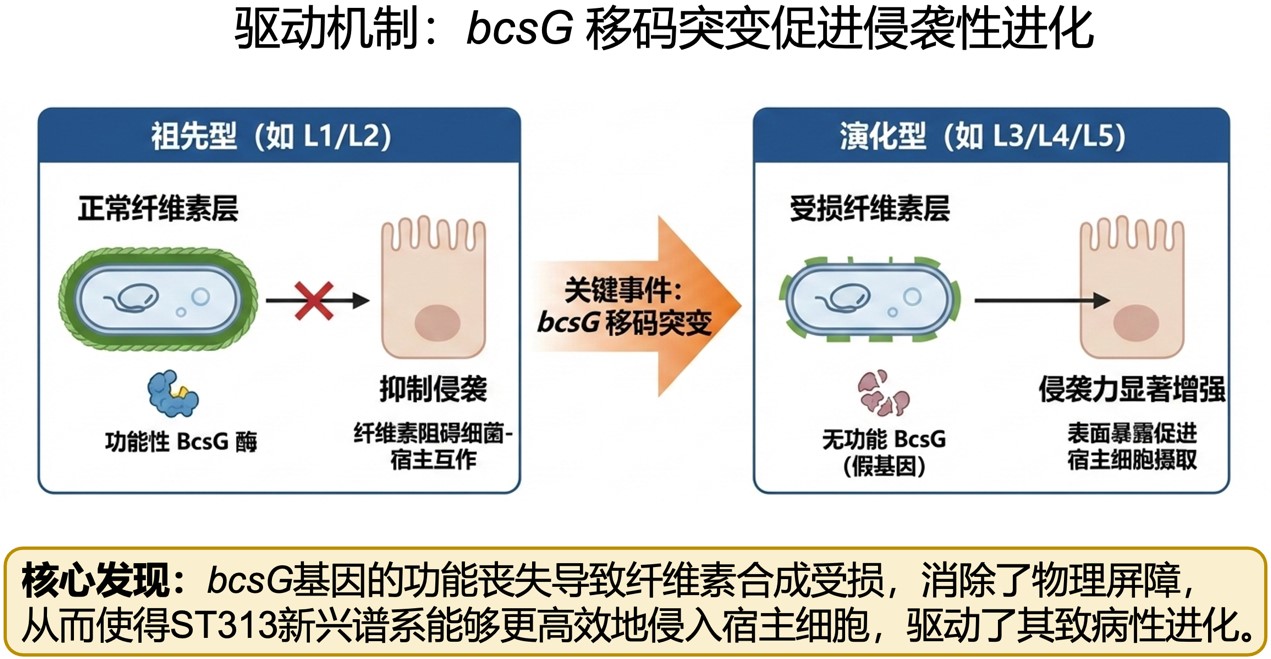
而比“更耐药”更值得注意的,是“更会侵袭”。研究发现,除了耐药谱的变化,ST313的持续演化还伴随着假基因积累以及部分功能基因受到正选择,这些遗传变化可能进一步增强其侵袭能力和竞争适应性。从分子层面,研究揭示了BcsG等相关基因的退化,介导病原菌表面的分子重构,导致对靶宿主细胞更强的侵袭性以及更强的胞内存活能力。正是这种分子层面的微小变化(功能基因丢失)让沙门菌获得了进化上竞争优势,驱动新的“低耐药”克隆成为高侵袭性菌型,进而导致竞争性取代。
总体而言,这项研究通过迄今最系统的iNTS与ST313基因组图谱,揭示了一个正在全球尺度上持续演化的高风险病原体:它一方面在现代人口流动背景下不断扩散,另一方面又在抗菌药物压力和可移动遗传元件的推动下,悄然完成谱系更新与适应性重塑。论文共同第一作者为国科大杭高院贾程皓和浙江大学周海洋,乐敏为通讯作者。本研究也得到了李艳教授(浙江大学),蓝乐夫研究员(国科大杭州高等研究院)等人的大力支持。
