
通讯单位:1) 国科大杭州高等研究院化学和材料学院 2) 中国科学院上海有机化学研究所
论文DOI:10.1002/ange.202209087
一、全文速览
国科大杭州高等研究院、中科院上海有机所汤文军课题组近期通过膦配体和催化模式的创新发展了一个高效的钯催化氮杂芳基卤代物和杂环烯烃的分子间Heck反应,实现了一系列手性α-杂芳基取代杂环的高效构建。该方法具有底物普适性好、官能团兼容性佳和反应条件温和等特征,研究团队基于该方法完成了假木贼碱和(S)-尼古丁及其类似物的高效合成。
二、背景介绍
手性α-杂芳基取代的杂环结构广泛地存在于众多具有重要生物活性天然产物和药物分子结构中。例如,(S)-尼古丁是烟草中针对中枢神经系统疾病的主要成分,生物碱Harmicine具有解痉、解热和抗癌特性,而缬苯那嗪已被批准用于治疗迟发性运动障碍(图1)。

▲图1 含有手性α-杂芳基取代杂环片段的天然产物和生物分子
手性α-杂芳基取代的杂环合成一直是不对称合成和药物研究领域中具有挑战性的研究课题。从结构上分析,氮杂芳基卤代物和杂环烯烃的分子间Heck反应是合成这类手性片段最直接和实用的方法之一。近年来手性催化剂和催化体系的发展有效促进了不对称Heck反应。国内外多个研究小组如Hayashi、Tietze、Pfaltz、侯雪龙、周建荣等在不对称 Heck 反应构建手性 α-芳基取代杂环领域做出了突出贡献。然而氮杂芳基卤化物和杂环烯烃的不对称Heck反应一直是一个悬而未决的难题,其中的关键原因在于氮杂芳基化合物特别是吡啶类结构与过渡金属的强配位性导致催化剂失活,因此至今成功的例子鲜有报道(图2)。

▲图2,氮杂芳基卤化物与杂环烯烃的不对称Heck反应
三、本文亮点
1)研究团队设计并发展了大位阻P, P=O配体,有效解决了钯催化Heck反应对氮杂芳基卤化物的兼容性问题,显著提高了反应的活性和底物普适性。
2)研究团队采用大位阻、富电性的DTBM-SEGPHOS为手性配体并结合阳离子钯催化模式实现了吡啶类底物的高对映选择性Heck反应,并利用不对称Heck反应实现了(S)-尼古丁及其类似物的高效合成。
3)该方法具有底物普适性好、官能团兼容性佳,反应条件温和等特点,为手性α-杂芳基取代杂环化合物提供了一个高效简洁的合成策略。
四、图文解析
研究团队首先以3-溴吡啶 (1a) 和 3,4-二氢吡啶-1(2H)-羧酸叔丁酯 (2a) 为模版底物展开研究。由于Heck反应产物通常是烯烃迁移混合物,因此Heck反应粗产物直接氢化形成单一的还原产物3a便于产物表征和产率计算。研究团队初步筛选了具有代表性的商业化单膦配体,例如SPhos, QPhos, BI-DIME (L1)等以及双膦配体包括Me-DuPhos, Ph-BPE, BINAP等和基于苯并氧杂五元膦烷的配体L5-L8,所有反应均未能得到目标产物,这表明含有吡啶基官能团的Heck反应确实具有挑战性。令人欣喜的是,当使用P, P=O 型配体L4时,反应以31%的产率得到产物3a,这表明P, P=O型配体在对含有吡啶基官能团的耐受方面具有显著的促进效果。研究团队进一步优化了P,P=O型配体结构和反应条件,发现L9是最佳配体,MeOH和Pd(OAc)2分别是最优溶剂和催化剂前体(图3)。


▲图3,条件优化
在最优条件下,研究团队首先对Heck反应的底物普适性进行了考察(图4)。3-溴吡啶和含有富电子取代基的3-溴吡啶与3, 4-二氢吡啶-1(2H)-羧酸叔丁酯反应可以以良好的收率(75-80%)生成了相应的产物3a-c。对于间位取代的吡啶底物,反应产率略有降低 (72%)。此外,使用邻位、间位二取代的3-溴吡啶为底物可以以中等收率得到相应的产物3e-g。值得注意的是,该方法能兼容大位阻取代基和多样取代形式的底物,例如,当使用2, 3-二氢吡喃作为烯烃底物时,间溴吡啶(3k)与其他氮杂芳基溴化物,包括邻位取代 (3l-n)、间位取代 (3o) 或邻、间二取代 (3p-3r) 以及含多个杂原子(3s)等底物均能以65-87%的收率得到相应的产物。同样地,2,3-二氢-1H-吡咯-1-羧酸叔丁酯也可以和吡啶基、喹啉基和嘧啶基溴化物发生Heck反应,以中等到良好的收率(65-88%)转化成相应的产物3u-3ad。此外,研究团队还探究了2, 3-二氢呋喃和氮杂芳基溴化物(3ae-3ap)之间的Heck反应,其中含有Me、F、CF3和CF2H等取代基的吡啶底物以及基于喹啉、嘧啶、异噻唑和吲唑的杂芳基溴代底物均可成功发生反应,彰显了该方法学的普适性和实用性。

▲图4,溴代氮杂芳环与杂环烯烃的Heck反应
为了阐明 P, P=O 型配体 L9 在促进氮杂芳基卤化物的 Heck 反应的作用机制,研究团队制备了[Pd(L9)Cl2]络合物,并利用 SambVca 2.1 Web 程序对 [Pd(L9)Cl2]与之前报道的[Pd(L11)Cl2]在位阻方面进行了分析(图5b)。结果表明具有蒽基的 [Pd(L9)Cl2] 络合物 (% Vbur = 57.6) 比带有二甲氨基的 [Pd(L11)Cl2] 络合物 (% Vbur = 51.1) 具有更加拥挤的催化口袋,从而更有效地抑制了与氮杂芳环的配位,并提高了钯催化剂的稳定性。同时,受益于P = O 的弱配位性,在氧化加成后 Pd(II) 配合物的P = O所占据的配位位点易于释放,促进了烯烃底物与钯中心的配位以确保后续迁移插入过程顺利进行。

▲图5. 配体结构-反应性分析
研究团队进一步研究了相应的不对称Heck反应。当使用手性P、P=O 配体时,产物的对映选择性较差。这可能是由于烯烃配位导致Pd中心与P = O基团的配位解离,在迁移插入过程中失去了有效的对映选择性控制。研究团队后续提出使用手性双膦配体和巧妙利用阳离子催化反应途径可能有利于提高反应的对映选择性(图 7a)。 研究团队通过对几种代表性手性膦配体进行系统考察后,发现采用(S)-DTBM-SEGPHOS作为配体时,产物4a的对映选择性高达96% ee(图6)。在最优条件下,研究团队对氮杂芳基卤化物和2,3-二氢呋喃间的不对称Heck反应的底物普适性展开研究。邻位富电子取代的3-溴吡啶以良好的收率(61-80%)和优异的ee(96-97%)生成了产物4b-e,同样地,具有吸电子取代基(2-F、2-CHF2、2-CF3、2-COOEt、2-COMe、2-吡啶基)的产物4f-k也以中等产率(52-65%)成功合成,并具有优异的对映选择性 (91-98%)。此外,该方法对于邻位含有取代基的溴代物同样具有良好的耐受性,以优异对映选择性合成了目标产物4z和4ac。研究团队还考察了含有各种官能团的 4-溴吡啶的底物普适性,所考察的底物都能以良好的收率 (63-70%) 和高ee(95-98%)转化为产物,包括4ad、4ae、4af。值得注意的是,该方法对于含多个杂原子的杂芳基结构如吡嗪(4ah)、嘧啶(4aj)、异噻唑(4ak)和吡唑(4ai,4al)均具有良好的兼容性,所得产物均具有良好的对映选择性 (86-97%)。除了溴代氮杂芳基,2-溴噻吩和3-溴苯并噻吩也可以作为该反应的亲电试剂,并以中等收率和良好的对映选择性合成相应产物4am和4an。此外,研究团队还探究了N-Boc-2,3-二氢吡咯作为杂环烯烃的不对称Heck反应,通过与溴代吡啶和溴代异喹啉反应得到了高光学活性产物 4ao和 4ap。
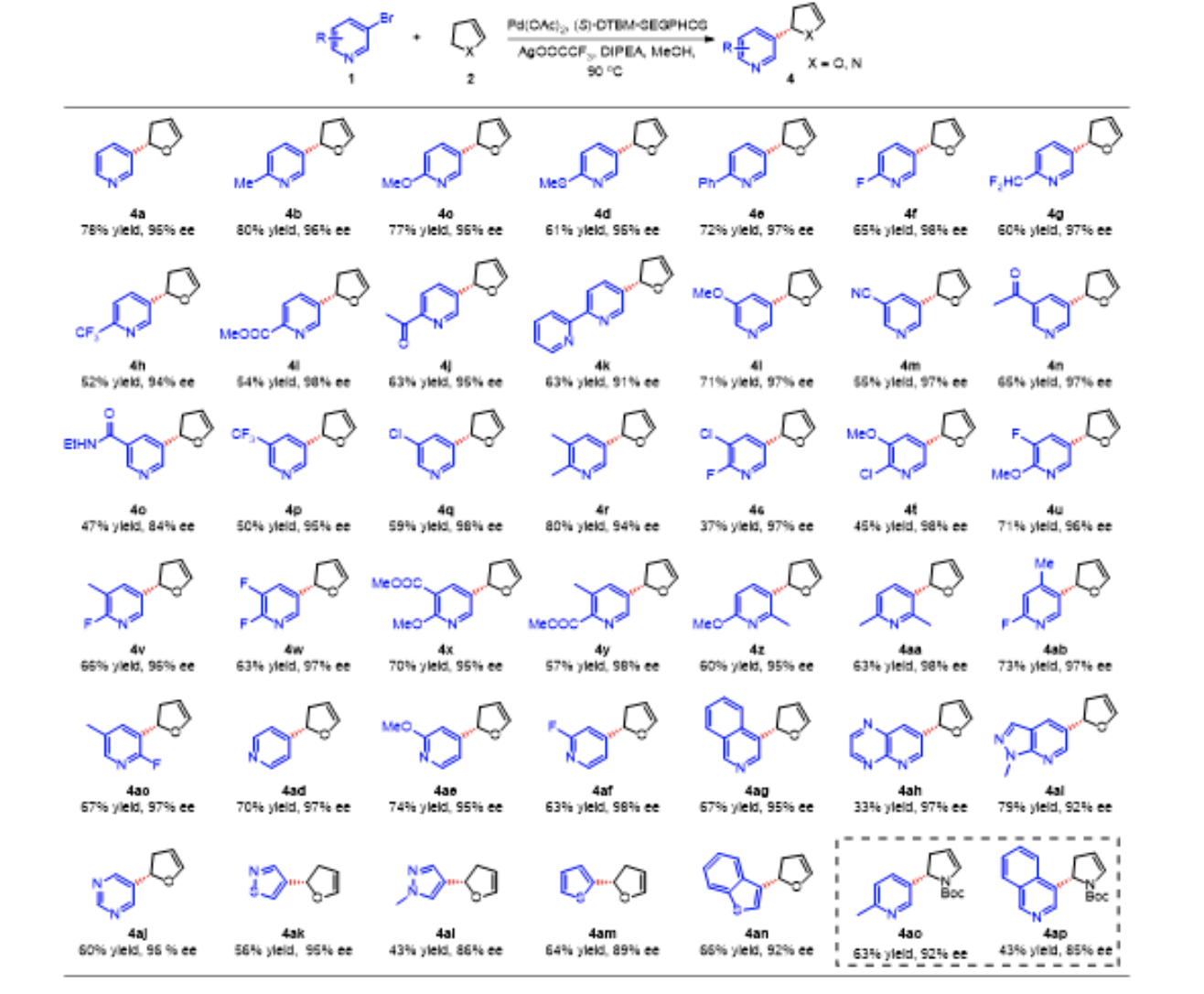
▲图6,溴代氮杂芳环参与的不对称Heck反应
研究团队提出了以(S)-DTBM-SEGPHOS为手性配体的不对称Heck反应的立体模型。在CF3COOAg作用下,氧化加成络合物中的溴原子从钯上解离下来,形成具有一个空配位点的阳离子 Pd 物种,有利于随后的烯烃配位。由于 (S)-DTBM-SEGPHOS 配位的 Pd 催化剂的空间环境,研究团队认为二氢呋喃等环状烯烃的 Re 面与 Pd 中心的配位更有利。此外,烯烃位于象限图的右下区域,基本上避免了与右上区域的配体芳基部分的空间排斥作用。随后经迁移插入生成相应的与氧原子相邻的三级立体中心。另外,研究团队指出目前该不对称催化体系对二氢吡喃或四氢吡啶类底物的反应效率还有待提高,研究团队推测六元环底物结构的空间位阻效应和半椅式构象不利于Heck反应的发生(图7b)。

▲图7,不对称 Heck 反应及立体化学模型
为了验证该方法的实用价值,研究团队使用Pd/L9 作为催化剂,在克级规模下实现了3-溴吡啶 (1a) 和3, 4-二氢吡啶-1(2H)-羧酸叔丁酯(2a)的分子间 Heck 反应,经氢化后以70%的产率得到产物3a,随后使用CF3COOH脱除3a中的N-Boc 保护基团顺利地合成了假木贼碱(anabasine,图8a)。此外,研究团队完成了3-溴吡啶 (1a) 与2, 3-二氢呋喃(2d)克级规模的不对称 Heck 反应,以74%的收率和96% ee生成了产物4a。随后经过钯/碳氢化,以99%的产率、96%的ee得到化合物(S)-3u(图8b)。(S)-Nicotine 是一种有效的多巴胺受体激动剂,研究团队使用Pd(OAc)2和(S)-DTBM-SEGPHOS催化剂,实现了3-溴吡啶(1a)与2c的不对称Heck反应,以 64% 的收率和 94% ee 合成了手性产物 6。随后经催化氢化和LiAlH4还原,顺利得到高光学活性的尼古丁7(图8c)。

▲图8,合成应用
五、总结与展望
国科大杭州高等研究院化学和材料学院、中科院上海有机所汤文军课题组通过发展大位阻P, P=O配体和新的催化模式成功实现了氮杂芳基卤化物的Heck反应,高效合成了一系列手性α-杂芳基取代的杂环结构。通过采用大位阻、富电性的DTBM-SEGPHOS为手性配体和阳离子钯催化反应途径实现了吡啶类底物的首次高对映选择性Heck反应。该方法具有底物普适性好、官能团兼容性佳和反应条件温和等特点,为具有α-杂芳基取代杂环骨架的生物活性分子提供了简洁实用的合成策略,并有望应用于新药研发和化学工艺开发中。
该工作得到了国家重点研发计划、国家自然科学基金委、中科院、上海有机所和广东省重点领域研发计划的大力资助。
六、课题组介绍
汤文军研究员简介

汤文军博士,研究员,1995年毕业于华东理工大学精细化工系,1998年于中国科学院上海有机化学研究所获硕士学位(导师:马大为院士);2003年于美国宾夕法尼亚州立大学获博士学位(导师:张绪穆教授)。2003-2005 年在美国Scripps研究所从事博士后研究(导师:K. C. Nicolaou教授),2005-2009年在美国Boehringer Ingelheim药物工艺部门任高级科学家 (Senior Scientist),2009-2011年任首席科学家 (Principal Scientist),2011年7月起任中科院上海有机化学研究所研究员、课题组长;同时兼任上海科技大学物质学院教授、博导、杭州高等研究院首席教授及华东理工大学药学院博导,2015年获中国均相催化青年奖,2017年获国家杰出青年基金资助。2018年入选科技部中青年科技创新领军人才。2019年获药明康德生命化学研究奖学者奖。
过渡金属催化是当代有机合成化学最活跃的研究领域之一。在过渡金属参与的催化反应中,过渡金属催化剂是整个反应过程的关键,而配体的特性包括电子效应,立体效应,和空间效应直接影响了催化剂的活性和选择性。手性膦配体的发展极大地推动了整个不对称催化领域。在过去的几年中,我们研究小组设计和发展了一系列有显著结构特征的P-手性膦配体在大位阻偶联、不对称偶联、不对称环化、以及不对称氢化中表现出优异的效率,为复杂天然产物和药物的高效合成提供了实用、绿色的催化反应方法学。
利用我们研究小组发展的合成方法学,开展对一些有重要生理活性天然产物的高效全合成及规模化制备,并进行相关药化研究。
发展能工业化的、高效、经济的新反应或合成策略,研究对药物或农药分子的高效合成工艺。
课题组诚招有理想和抱负的博士后,有意向者请联系汤文军研究员。
http://wenjuntang.sioc.ac.cn/